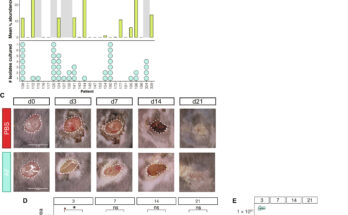
inhibits glucose metabolism. <b>b</b>, <b>c</b> Insulin secretion (<b>b</b>) and insulin content (<b>c</b>) in LG-cells and HG-cells cultured for 48 h ± 10 mM mannoheptulose (MANNO) and then stimulated with 2 mM or 20 mM glucose. Mannoheptulose was omitted during the assay (<i>n</i> = 3 biologically independent experiments). <b>d</b> Oxygen-consumption rate (OCR) in LG-cells and HG-cells cultured for 48 h ± 10 mM MANNO. OCR was recorded at 2 mM glucose and after sequential addition of 20 mM glucose (20 G), 1 μM oligomycin (Oligo) and 0.5 μM rotenone + 0.5 μM antimycin A (Rot + Ant). Data are expressed as the percentage change from baseline (2 mM glucose); <i>n</i> = 10 biologically independent experiments per group. <b>e</b> Percentage change in OCR when glucose was raised from 2 to 20 mM (20 G), ATP-linked OCR (Oligo), OCR required to maintain the mitochondrial leak (Rot + Ant) and non-mitochondrial OCR (non-mito); <i>n</i> = 10 biologically independent experiments per group. Same data as in (<b>d</b>). <b>f</b>, <b>g</b> mRNA levels of the indicated genes involved in glycolytic (<b>f</b>) and mitochondrial (<b>g</b>) metabolism as assessed by qPCR in LG-cells and HG-cells cultured for 48 h ± 10 mM mannoheptulose (<i>Pdk1, Idh2</i> and <i>Ndufs8</i>, <i>n</i> = 6 biologically independent experiments; <i>Ndufa4</i>, <i>n</i> = 4 biologically independent experiments; <i>Pfkl</i>, <i>Pfkfb3</i>, <i>Eno1</i>, <i>Sdha</i> and <i>Mdh2</i>, <i>n</i> = 3 biologically independent experiments; <i>Aldob</i>, <i>n</i> = 6 biologically independent experiments for LG and HG but <i>n</i> = 5 for LG + MANNO and HG + MANNO; <i>Ndufs8</i>, <i>n</i> = 6 biologically independent experiments for LG but <i>n</i> = 3 for LG + MANNO, HG and HG + MANNO). All panels show individual data points and mean ± s.e.m. *<i>P</i> < 0.05, **<i>P</i> < 0.01, ***<i>P</i> < 0.001; two-tailed unpaired Student’s <i>t</i> test. LG-cells (black), HG-cells (red), LG-cells + mannoheptulose (blue), HG-cells + mannoheptulose (orange). Credit: <i>Nature Communications</i> (2022). DOI: 10.1038/s41467-022-34095-x")
Glucose metabolites (chemicals produced when glucose is broken down by cells), rather than glucose itself, have been discovered to be key to the progression of type 2 diabetes. In diabetes, the pancreatic beta-cells do not release enough of the hormone insulin, which lowers blood glucose levels. This is because a glucose metabolite damages pancreatic beta-cell function.
An estimated 415 million people globally are living with diabetes. With nearly 5 million people diagnosed with the condition in the UK, it costs the NHS some £10 billion each year. Around 90% of cases are type 2 diabetes (T2D), which is characterized by the failure of pancreatic beta-cells to produce insulin, resulting in chronically elevated blood glucose.
T2D normally presents in later adult life, and by the time of diagnosis, as much as 50% of beta cell function has been lost. While researchers have known for some time that chronically elevated blood sugar (hyperglycemia) leads to a progressive decline in beta-cell function, what exactly causes beta-cell failure in T2D has remained unclear.
Now a new study led by Dr. Elizabeth Haythorne and Professor Frances Ashcroft of the Department of Physiology, Anatomy and Genetics at the University of Oxford has revealed how chronic hyperglycemia causes beta-cell failure.
Using both an animal model of diabetes and beta-cells cultured at high glucose, they showed, for the first time, that glucose metabolism, rather than glucose itself, is what drives the failure of beta-cells to release insulin in T2D. Importantly, they also demonstrated that beta-cell failure caused by chronic hyperglycemia can be prevented by slowing the rate of glucose metabolism.
Professor Ashcroft said, “This suggests a potential way in which the decline in beta-cell function in T2D might be slowed or prevented.”
The blood glucose concentration is controlled within narrow limits. When blood glucose is too low for more than few minutes, consciousness is rapidly lost because the brain is starved of fuel. Chronic elevation of the blood glucose concentration is also dangerous, as it gives rise to the serious complications found in poorly controlled diabetes such as retinopathy, nephropathy, peripheral neuropathy, and cardiac disease.
Insulin, released from pancreatic beta-cells when blood glucose levels rise, is the only hormone that can lower the blood glucose concentration and diabetes (high blood glucose) results if insulin secretion is insufficient. In T2D, the beta-cells are still present (unlike T1D), but they have a reduced insulin content and the coupling between glucose and insulin release is impaired.
Previous work from the Oxford team has shown that chronic hyperglycemia damages the ability of the beta-cell to produce insulin and to release it when blood glucose levels rise. This suggested that prolonged hyperglycemia sets off a vicious spiral in which an increase in blood glucose leads to beta-cell damage and less insulin secretion—which causes an even greater increase in blood glucose and a further decline in beta-cell function.
Dr. Haythorne said, “We realized that we next needed to understand how glucose damages beta-cell function, so we can think about how we might stop it and so slow the seemingly inexorable decline in beta-cell function in T2D.”
The team’s new study is important because it shows that a breakdown product of glucose metabolism, rather than glucose itself, is what causes the failure of beta-cells to release insulin in diabetes. High blood glucose levels cause an increased rate of glucose metabolism in the beta-cell which leads to a metabolic bottleneck and the pooling of upstream metabolites.
These metabolites switch off the insulin gene, so less insulin is made, as well as switching off numerous genes involved in metabolism and stimulus-secretion coupling. Consequently, the beta-cells become glucose blind and no longer respond to changes in blood glucose with insulin secretion.
Crucially, the team found that blocking an enzyme called glucokinase, which regulates the first step in glucose metabolism, could prevent the gene changes taking place and maintain glucose-stimulated insulin secretion even in the presence of chronic hyperglycemia.
Professor Ashcroft said, “This is potentially a useful way to try to prevent beta-cell decline in diabetes. Because glucose metabolism normally stimulates insulin secretion, it was previously hypothesized that increasing glucose metabolism would enhance insulin secretion in T2D and glucokinase activators were trialed, with varying results.”
“Our data suggests that glucokinase activators could have an adverse effect and, somewhat counter-intuitively, that a glucokinase inhibitor might be a better strategy to treat T2D. Of course, it would be important to reduce glucose flux in T2D to that found in people without diabetes—and no further.”
“But there is a very long way to go before we can tell if this approach would be useful for treating beta-cell decline in T2D. In the meantime, the key message from our study if you have type 2 diabetes is that it is important to keep your blood glucose well controlled.”
The research was published in Nature Communications.
Elizabeth Haythorne et al, Altered glycolysis triggers impaired mitochondrial metabolism and mTORC1 activation in diabetic β-cells, Nature Communications (2022). DOI: 10.1038/s41467-022-34095-x
Citation:
Study uncovers key cause of type 2 diabetes (2022, November 14)
retrieved 14 November 2022
from https://medicalxpress.com/news/2022-11-uncovers-key-diabetes.html
This document is subject to copyright. Apart from any fair dealing for the purpose of private study or research, no
part may be reproduced without the written permission. The content is provided for information purposes only.
Source link